
Soluble guanylate cyclase agonists: Have we found a new “new drug” for heart failure?
| Take Home Messages |
|---|
|
Introduction
Heart failure (HF) is a global disease with multiple aetiologies. It remains a condition associated with a significant morbidity and mortality. Although substantial advances have been made with regards to medical and device-based therapies over the last few decades, there is a need for further advances in therapy to reduce its burden on the health of populations across the world. An emerging therapeutic target for HF is the nitric oxide-soluble guanylate cyclase-cyclic guanosine monophosphate (NO-sGC-cGMP) pathway, with newer agents showing some promising results via action through this pathway. Besides early trials on animals, there have already been a few clinical trials investigating the use of sGC agonists in the context of HF. This includes a range of novel agents such as cinaciguat, riociguat and vericiguat. In this review, the pharmacology of this class of agents is first discussed, followed by a review of the phase IIb and phase III trials undertaken so far.
Background
Heart failure
The global burden of HF cannot be overstated with an estimated 64.3 million people currently living with the condition worldwide1. There are an estimated 900,000 people with HF in the UK and the disorder is associated with 5% of all emergency hospital admissions in adults2.
There have been significant advances in the management of HF over the last few decades, especially for patients with reduced ejection fraction. The more recent advances have included device therapy in the form of cardiac resynchronisation therapy and newer agents such as sacubitril/valsartan3, dapagliflozin4 and empagliflozin5 which have shown improved patient outcomes. It is, however, a condition which continues to have a five-fold increased risk of death6 and, disappointingly, there remains no prognostically beneficial treatment for patients with heart failure with preserved ejection fraction (HFpEF)7. The new guidelines for heart failure have introduced the concept of heart failure with mid-range ejection fraction (HFmrEF) and there does appear to be some data of effectiveness of some of the newer agents, such as sacubitril/valsartan, within this group8. Nevertheless, further advances must be made to improve our knowledge of the condition and develop therapies that can improve the mortality and indeed, the quality of life, of our patients.
The NO-sGC-cGMP Pathway
It is known that patients with HF have dysregulation of physiological pathways9. One of these pathways involves nitric oxide (NO), which is at the beginning of several essential physiological processes. An extensive explanation of the role of NO is beyond this review and can be found elsewhere10–12. In brief, NO is generated within the endothelium and diffuses into smooth muscle cells. Following entry, it binds to soluble guanylate cyclase (sGC) which leads to the synthesis of the second messenger cyclic guanosine monophosphate (cGMP), which eventually leads to vasodilation and reduces smooth muscle proliferation, leukocyte recruitment and platelet aggregation. cGMP is essential for the normal functioning of the cardiovascular system with a dysfunction of this system implicated in coronary microvascular dysfunction, vascular stiffness, myocardial fibrosis and dysfunction13.
As a result of the multiple mechanisms by which this pathway impacts physiological processes, it has been felt that it could be targeted to improve outcomes in patients with multiple cardiopulmonary diseases including pulmonary hypertension, arterial hypertension, atherosclerosis and HF. Figure 1 describes some of the key steps in the pathway and the important targets of drug therapy. In HF there is reduced bioavailability of NO as well as relatively increased forms of oxidised sGC which are insensitive to NO14. An increased activation of the NO-sGC-cGMP pathway in patients with HF could reduce progression of ventricular hypertrophy and fibrosis as well as reduce left and right ventricular afterload13. This increased pathway activation could be coordinated by administration of organic nitrate compounds
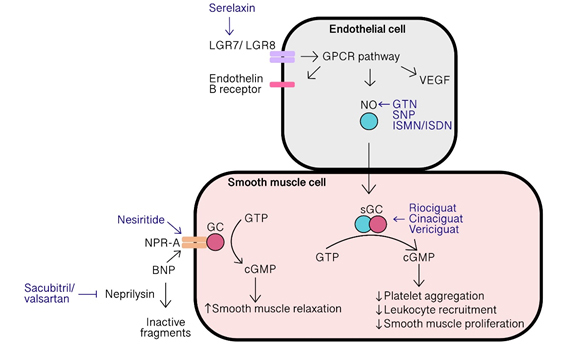
Figure 1: The NO-sGC-cGMP pathway and the targets for key drugs used in heart failure (original figure, also see Refs 15,32,33). LGR, leucine-rich repeat G-protein coupled receptor; GPCR, G protein coupled receptor; VEGF, vascular endothelial growth factor; NO, nitric oxide; GTN, glyceryl trinitrate; SNP, sodium nitroprusside; ISMN, isosorbide mononitrate; ISDN, isosorbide dinitrate; sGC, soluble guanylate cyclase; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate; GC, guanylate cyclase; NPR-A, natriuretic peptide receptor A; BNP, B-type natriuretic peptide.
Organic nitrates such as glyceryl trinitrate (GTN), sodium nitroprusside (SNP) and isosorbide mononitrate (ISMN) act as NO donors and thereby cause vasodilation with differing effects on veins and arteries based on dosing15. These compounds have been used in acute heart failure for many years but there is little evidence for their use to improve mortality, although a combination of isosorbide dinitrate and hydralazine has been shown to be beneficial in addition to standard therapy in the African-American group(16).
Table 1 describes some of the trials looking at vasodilators in heart failure. More recently the GALACTIC randomised clinical trial(17) showed that early intensive vasodilation using a combination of vasodilators including nitrates, hydralazine, angiotensin-converting enzyme inhibitor, angiotensin receptor blocker and sacubitril/valsartan, in patients with acute heart failure does not improve all-cause mortality or rehospitalisation.
| Table 1. Summary of vasodilators used in HF | ||
|---|---|---|
| Drug, Trial | Design | Outcome |
| ISDN34 | Randomised, open-label. IV ISDN vs. furosemide/morphine | No significant difference between groups in PaO2/FiO2 ratio |
| ISDN35 | Randomised, open-label. IV ISDN vs. IV furosemide | Reduced SBP, peripheral resistance, pulmonary vascular pressures and LV filling pressures acutely |
| ISDN36 | Randomised, single-blind. IV ISDN vs. IV furosemide vs. IV hydralazine vs. IV prenalterol | Reduced LV filling pressures acutely |
| Nitroglycerin, Nesirtide37 | Randomised, double-blind. IV nitroglycerin vs IV nesiritide vs placebo | No significant difference in mean PCWP. (nitroglycerin). Reduced PCWP and dyspnoea (nesiritide) |
| ISDN & hydralazine16 | Randomised, placebo controlled, double-blind. Oral ISDN & hydralazine vs. placebo | Improved weighted composite of all-cause mortality or first hospitalisation for HF over 18 months, and change in quality of life at 6 months |
| Nesiritide38 | Randomised, double-blind, placebo controlled. IV nesiritide vs placebo | No significant improvement in dyspnoea, 30-day mortality or rehospitalisation |
| Serelaxin39 | Randomised, double-blind, placebo-controlled | Serelaxin improved dyspnoea (visual analogue scale area under the curve) to day 5. No change in CV death or readmission for HF or renal failure at 60 days. |
| Abbreviations: ISDN, isosorbide dinitrate; IV, intravenous; SBP, systolic blood pressure; LV, left ventricle; PCWP, pulmonary capillary wedge pressure; CV, cardiovascular | ||
One of the limitations of the NO donor agents is that patients with cardiovascular disease have often developed resistance to NO such that platelets and arteries do not respond as well to the vasodilatory and anti-aggregatory effects of NO18. Furthermore, there are some cytotoxic effects of NO through other pathways19. To overcome these drawbacks, and since most of the beneficial effects of NO are mediated through the NO-sGC-cGMP pathway, this has now become a promising target for novel therapeutic agents. This includes two types of sGC agonists: sGC stimulators, which sensitise sGC to low levels of NO; and sGC activators, which cause the activation of sGC20. It is the latter type, or agents with mixed properties of stimulators and activators, that have been explored more thoroughly in HF (Table 2). One of the key benefits of these agents as opposed to the NO donors is that the latter are often associated with dose-dependent tolerance and reduced effectiveness whereas the sGC agonists don’t appear to have this issue15.
| Table 2. Summary of key trials of sGC agonists | ||||
|---|---|---|---|---|
| Drug, Trial | Design | HF setting | Primary endpoint | Result |
| Cinaciguat21 | Phase I, non-randomised, uncontrolled, unblinded, proof of concept | Acute decompensated | Change in PCWP at 6 hours | Significantly reduced |
| Cinaciguat22 | Phase IIb, randomised, double-blind, placebo-controlled | Acute decompensated | Change in PCWP at 8 hours | Significantly reduced |
| Riociguat23 | Phase IIa, randomised, double-blind, placebo-controlled, parallel group | Stable | Change in mPAP at 6 hours | No change |
| Vericiguat25 | Phase IIb, randomised, double-blind, placebo-controlled | Recent decompensation | NT-proBNP | No change |
| Vericiguat26 | Phase IIb, randomised, double-blind, placebo-controlled | Recent decompensation | NT-proBNP, LA volume | No change in either |
| Vericiguat30 | Phase III, randomised, double-blind, placebo-controlled, | Recent decompensation | Composite of CV death and first HF hospitalisation | Significantly reduced |
Evidence for sGC agonists
Cinaciguat
Cinaciguat is a novel sGC activator for which there have been a phase I and a phase IIb study looking at its use in acute decompensated HF. In pre-clinical studies it has been shown to preferentially act on sGC that is resistant to the effects of NO and can induce vasodilatation in diseased vessels20,21.
The first clinical study of cinaciguat, published in 2009, assessed its intravenous use in patients who were admitted with an acute decompensation of HF21. Although this was a non-randomised, uncontrolled, unblinded, proof of concept and phase I study, it did demonstrate some promising results.
A dose of 100 µg/h cinaciguat was chosen as a starting dose for the second part of the study and this was adjusted based on haemodynamic response. At the end of the 6-hour infusion, there was a significant drop in mean PCWP compared to baseline (17.2 ± 5.2 mmHg vs. 24.1 ± 5.4 mmHg, p<0.0001), with 90% of patients responding to the infusion following 6 hours. There was also a significant 40% increase in cardiac output and a significant improvement in dyspnoea scores in 26 out of the 30 participants at the end of the 6-hour infusion.
Whilst the results of the study were consistent with its expected vasodilatory effects, it needs to be noted that this study had several limitations given its methodology as it was a non-randomised, uncontrolled and unblinded trial. It’s also important to note that the cost of offloading the PCWP by 7 mmHg was a 14 mmHg drop in systolic blood pressure (SBP) which could have a significant impact on the drug’s use in the real world.
Cinaciguat has since been tested in the first placebo-controlled phase IIb study looking at sGC activators22 in which 139 patients admitted with acute decompensated HF, with a PCWP ≥18 mmHg, left ventricular ejection fraction (LVEF) <40% and a need for haemodynamic monitoring, were randomised to intravenous cinaciguat or placebo. The primary outcome was change in PCWP from baseline to 8 hours. Cinaciguat had similar haemodynamic effects as previously described with a placebo-adjusted fall in PCWP of 4.0 mmHg at a cost of a placebo-adjusted fall in SBP of 16.6 mmHg. Ventricular tachycardia was also seen in 6 of the patients in the treatment arm. There was no significant difference in 30-day mortality between the two groups.
Again, there were important limitations to this study. Firstly, there was a lack of control over other therapeutic agents being used, as these were not recorded. The study was also stopped early due to an excess of hypotension in the cinaciguat group. The significant drop in blood pressure is likely to make the use of the drug in settings of acute decompensated HF very difficult. One of the key take home messages from this study may well be the difficulty in carrying out a trial in such a setting where a patient is diagnosed with acute decompensated heart failure as there are likely to be problems with time to treatment with the study drug, as well as the fact that this is such a heterogeneous group of patients. There is likely to have been a delay between the patient attending the emergency department to receiving cinaciguat or placebo (up to 48 hours as per trial protocol) during which time they may have received other therapy such as diuretics and dobutamine. This delay could have resulted in an underestimated effect of cinaciguat. Despite these limitations, the study does, however, pave the way for further advances using other sGC activators in HF.
Riociguat
Riociguat is an sGC stimulator with a dual action of sensitisation of sGC to endogenous NO as well as stimulating sGC directly. It has already been approved for use in chronic thromboembolic pulmonary hypertension (CTEPH), idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with connective tissue disease. It has also been investigated in patients with pulmonary hypertension associated with HFpEF in the DILATE-1 study, which did not meet its primary outcome23. This was a randomised, double-blind, placebo-controlled study in clinically stable patients on standard HF therapy with a LVEF >50% and had a mean pulmonary artery pressure (mPAP) ≥25mmHg and pulmonary capillary wedge pressure (PCWP) > 15mmHg at rest.
The primary outcome was change in mPAP from baseline compared to 6 hours post-dose alongside haemodynamic and echocardiographic parameters and any potential safety issues. There was no significant difference in mPAP or PCWP. There was, however, a significant increase in stroke volume and cardiac index in patients receiving the 2 mg dose. This study was limited by the small numbers in each arm and the fact that it was based on a single dose with no significant long-term assessment of the drug.
Although there was no improvement of the mPAP, the increase in stroke volume and cardiac index without an increase in PCWP suggests the drug may be beneficial in patients with diastolic dysfunction and further clinical trials looking at the long term, longitudinal effect of riociguat may prove helpful. It is, however, important to understand that the main therapy in patients with pulmonary hypertension secondary to left sided heart failure remains diuresis and there is still a lack of evidence for the use of vasodilators in such patients24.
Vericiguat
Vericiguat is an sGC stimulator with the most clinical evidence demonstrated so far amongst this class of drugs. SOCRATES-REDUCED25 was a phase IIb randomised trial that assessed the optimal dose and tolerability of vericiguat in patients with a LVEF <45% and a recent episode of worsening HF. The primary end-point was change in log-transformed value of N-terminal pro-B-type natriuretic peptide (NT-proBNP) at week 12. There was no significant difference in log-transformed NT-proBNP in the pooled vericiguat group compared to placebo, although exploratory secondary analyses of the primary end-point using linear regression modelling suggested a dose-response relationship with higher doses leading to greater drops in NT-proBNP.
Interestingly, vericiguat has also been tried in HFpEF with some curious results. In SOCRATES-PRESERVED26, 477 patients underwent a randomised, placebo-controlled, double-blind, dose-finding phase IIb study. The primary endpoints were change from baseline at 12 weeks in log-transformed NT-proBNP and left atrial (LA) volume. The participants all had symptomatic (NYHA class II-IV) worsening HF with a LVEF ≥ 45%, increased BNP/NT-proBNP levels and enlarged LA.
The results revealed no significant difference in either primary endpoint but did show a significant difference in patient-reported outcomes including the Kansas City Cardiomyopathy Question-Clinical Summary Score (KCCQ-CSS) and the EQ-5D, which were both measured during the study27. The authors did suggest a few reasons for the neutral outcome for their primary endpoint. Firstly, previous studies have demonstrated that NT-proBNP levels may initially rise, at least temporarily, before falling more than 3 months after therapy has been initiated28. Secondly, the participants in the study had relatively high NT-proBNP levels (median 1174 pg/ml) and very large LA volumes (mean 86 ml) suggesting an advanced stage of the HFpEF phenotype. As a result, it may be that the 12 week follow up could have been simply too short to see a significant impact on their primary end points. Interestingly, another trial assessing the impact of vericiguat in HFpEF, VITALITY-HFpEF29, failed to confirm the findings of patient reported outcomes with there being no significant difference in physical limitation scores of the KCCQ after 24 weeks therapy with vericiguat compared to placebo. There may be a few reasons for this: for example, the VITALITY-HFpEF groups had higher baseline KCCQ scores and were less symptomatic with fewer NYHA class III patients.
The most recent study looking at vericiguat in HF is the VICTORIA trial (30). In this randomised, double-blind, placebo-controlled, phase III trial, 5050 patients with chronic HF with NYHA class II-IV and LVEF of <45% were randomised to receive either vericiguat at a target dose of 10 mg daily or placebo, in addition to other guideline-directed medical therapy. The primary outcome was a composite including death from cardiovascular causes or first hospitalisation for HF.
The participants included all patients who had a recent decompensation of their HF, in the form of either a hospital admission for HF in the preceding 3 months, hospital admission for HF in the preceding 3-6 months or the requirement of intravenous diuretic therapy in the preceding 3 months. They all also had a raised BNP (>300 pg/ml in sinus rhythm, >500 pg/ml in atrial fibrillation) or NT-proBNP (>1000 pg/ml in sinus rhythm, >1600 pg/ml in atrial fibrillation). Exclusion criteria included a systolic blood pressure <100 mmHg, concurrent use of nitrates, sGC stimulators, phosphodiesterase type 5 inhibitors, intravenous inotropes or LV assist devices.
From the 5050 participants recruited, median follow up was 10.8 months, mean age was 67.3 years, 76.1% were male, mean LVEF was 28% and median NT-proBNP was 2816 pg/ml. The dropout rate was relatively high with 23.2% of participants having discontinued trial regimen after randomisation across the groups and 89.2% of the vericiguat group maintaining the target dose at 12 months.
The authors reported a significant reduction in the primary composite outcome with 35.5% in the vericiguat group compared to 38.5% in placebo group (hazard ratio[HR] 0.90, 95% confidence interval[CI] 0.82 - 0.98, p=0.02). This was driven by a statistically significant reduction in total hospitalisations (38.3% vs. 42.4%, HR 0.91, CI 0.84-0.99). However, there was only a borderline reduction in first hospitalisations (HR 0.90, 95% CI 0.81-1.00). Serious adverse events were reported in 32.8% of the vericiguat group compared to 34.8% in the placebo group. Symptomatic hypotension and syncope were not statistically different between the groups.
This appears to be a successful trial in suggesting that vericiguat may be considered as an additional therapy in patients with HF with a 10% reduced risk of death from cardiovascular causes or hospitalisation for HF. It is, however, important to note several points. Firstly, the primary outcome was a composite outcome which should always be interpreted with caution. When death from cardiovascular causes on its own is analysed, it was not statistically significant (16.4% vs. 17.5%, HR 0.93, CI 0.81-1.06). Thus, the composite end point was largely driven by hospitalisation for HF.
The authors commented on a number needed to treat (NNT) with vericiguat for 1 year to prevent cardiovascular death or hospitalisation for HF of 24 patients. This compares to a NNT of 21 patients for the same outcome with sacubitril/valsartan at 27 months (3). Although the end point for death from cardiovascular cause did not reach statistical significance, it’s important to also note that the patients in this study were a high-risk group. The patients had all had a recent decompensation within 3 months or 3-6 months. In PARADIGM-HF (3) and DAPA-HF (4), the proportion of patients with NYHA class III or IV symptoms was 25% and 32% respectively, in comparison to 41% in this study. The patients in this study also had higher median NT-proBNP levels compared to PARADIGM-HF and DAPA-HF (2816 pg/ml vs. 1608 pg/ml vs. 1437 pg/ml, respectively). Overall this makes these patients at higher risk which explains the increased mortality across both the placebo and treatment groups in this study compared to previous large trials. As a result, the event rate was high and therefore the median follow-up of 10.8 months was sufficient to show the composite end point. It may be that this was too short a time to prove a benefit in cardiovascular death and a longer duration of observation may have shown this.
Of note, although the study did meet its primary end-point, the numbers are not as convincing when you look beyond the p-value. A useful method of assessment of the robustness of a study is the use of the fragility index (FI)(31). For the primary end-point in this study, the FI calculates as 8, meaning that if only 8 patients from the treated group were transferred from a non-event to an event outcome then there would be no significant difference in the primary end-point. Given that the number of patients who were lost to follow up in the treatment arm in the study was 9, this puts the robustness of the study into doubt.
Looking at the sub-group analysis, it appears that the impact of the drug may vary based on baseline NT-proBNP level, which worryingly shows a trend towards harm in the top quartile of NT-proBNP subgroup of >5314 pg/ml (HR 1.16, CI 0.99-1.35) when compared to placebo. It may also have been useful to see the impact of the drug on NT-proBNP levels for the duration of the study in addition to other measures such as mPAP, PCWP and exercise testing.
Conclusions
Clearly there is great interest in the use of sGC agonists in the future management of HF. Until recently, we have only had small trials that show, at most, mildy promising results to suggest their scope to become the new “new drug” for HF. Vericiguat appears to have made some progress in providing evidence for its role in patients with reduced ejection fraction, albeit there is only one study that has so far shown a reduction in hospitalisation and mortality. More interestingly, it also appears to have shown some benefit in patients with HFpEF, at least regarding quality of life. Let alone whether this class of agents represent a possible new “new drug” for HF, could we indeed finally have a drug for patients with preserved ejection fraction? Unfortunately, VITALITY-HFpEF seems to suggest not. Larger, longitudinal studies with hard outcomes in HFpEF will likely be needed to answer this essential question.
The findings of GALACTIC seem to suggest that we should be moving away from the vasodilators in the management of heart failure. It is important to note, however, that this was in the context of acute decompensated heart failure as opposed to stabilised heart failure and there may yet be a way forward with the non-nitrate donor vasodilators such as the sGC agonists in the chronic HF.
Far more evidence is required before sGC agonists can be incorporated into routine care for our HF patients, who of course are a heterogenous group. It may be that the way forward is to look at specific cohorts of patients with HF rather than the broad categories of HFrEF vs HFmrEF vs HFpEF. Could sGC agonists, and indeed, other vasodilators be more effective in specific types and causes of heart failure?
References
- Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. 2020;22(8):1342–56.
- NICOR. National heart failure audit. 2019;
- McMurray JJV, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N Engl J Med. 2014;371(11):993–1004.
- McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2019;381(21):1995–2008.
- Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N Engl J Med. 2020;383(15):1413–24.
- Magnussen C, Niiranen TJ, Ojeda FM, Gianfagna F, Blankenberg S, Vartiainen E, et al. Sex-Specific Epidemiology of Heart Failure Risk and Mortality in Europe: Results From the BiomarCaRE Consortium. JACC Hear Fail. 2019;7(3):204–13.
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37:2129–200.
- Solomon SD, Vaduganathan M, L. Claggett B, Packer M, Zile M, Swedberg K, et al. Sacubitril/Valsartan across the Spectrum of Ejection Fraction in Heart Failure. Circulation. 2020;141(5):352–61.
- Packer M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol. 1992;20(1):248–54.
- Lowenstein CJ, Snyder SH. Nitric oxide, a novel biologic messenger. Cell. 1992;70(5):705–7.
- Murad F. Nitric Oxide and Cyclic GMP in Cell Signaling and Drug Development. N Engl J Med. 2006;355(19):2003–11.
- Förstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. Eur Heart J. 2012;33(7):829–37.
- Stasch JP, Pacher P, Evgenov O V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263–73.
- Münzel T, Genth-Zotz S, Hink U. Targeting heme-oxidized soluble guanylate cyclase: Solution for all cardiorenal problems in heart failure? Hypertension. 2007;49(5):974–6.
- Singh A, Laribi S, Teerlink JR, Mebazaa A. Agents with vasodilator properties in acute heart failure. Eur Heart J. 2017;38:317–25.
- Taylor AL, Ziesche S, Yancy C, Carson P, D’Agostino R, Ferdinand K, et al. Combination of Isosorbide Dinitrate and Hydralazine in Blacks with Heart Failure. N Engl J Med. 2004;351(20):2049–57.
- Kozhuharov N, Goudev A, Flores D, Maeder MT, Walter J, Shrestha S, et al. Effect of a Strategy of Comprehensive Vasodilation vs Usual Care on Mortality and Heart Failure Rehospitalization Among Patients With Acute Heart Failure The GALACTIC Randomized Clinical Trial. J Am Med Assoc. 2019;322(23):2292–302.